Organophosphorus chemistry
Dynamic resolution of P-Stereogenic phosphorus compounds. This unprecedented reaction enables easy construction of P-stereogenic phosphorus compounds (incl. phosphoramidites). It was run at kg scale and commercialized. It is based on an asymmetric version of the Appel conditions – I chose to look at this old reaction from the point of view of the unwanted by-product and realised that we could take advantage of its high selectivity to make much more desirable end-products. From initial promising results (publication P1) the project has matured to be a powerful and flexible method for the construction of a wide range of chiral phosphorus compounds (publication P2), which was patented (PCT/IB2005/002079 granted) and licensed for commercialization to Celtic Catalysts (our Campus Company set up for that purpose). We found interesting mechanisms and intermediates (Chem Commun, 2012, 48, 10040) and discovered fundamental insights into the rates of nucleophilic substitution at phosphonium centres notably: pentavalent transition states vs. pentavalent intermediates (Publication P3).
Selected Publications
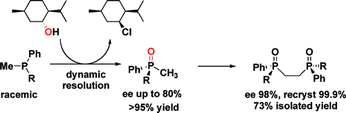
Synthesis of P-Stereogenic Phosphorus Compounds. Asymmetric Oxidation of Phosphines under Appel Conditions
Racemic phosphines are converted into enantioenriched phosphine oxides via a synthetically simple, but theoretically interesting, oxidation procedure in good enantiomeric excess (up to 80%) and excellent yields (>95%). These phosphine oxides can be oxidatively coupled to provide easy access to enantiopure DiPAMPO analogues. Particularly attractive aspects of this procedure are the operational simplicity and the low cost required to synthesize these high value compounds.
Enda Bergin , Cormac T. O'Connor, Shane B. Robinson, Eoghan M. McGarrigle, Colm P. O'Mahony, Declan G. Gilheany (2007) Journal of the American Chemical Society, 129 (31), 9566-9567 (link to paper)
Racemic phosphines are converted into enantioenriched phosphine oxides via a synthetically simple, but theoretically interesting, oxidation procedure in good enantiomeric excess (up to 80%) and excellent yields (>95%). These phosphine oxides can be oxidatively coupled to provide easy access to enantiopure DiPAMPO analogues. Particularly attractive aspects of this procedure are the operational simplicity and the low cost required to synthesize these high value compounds.
Enda Bergin , Cormac T. O'Connor, Shane B. Robinson, Eoghan M. McGarrigle, Colm P. O'Mahony, Declan G. Gilheany (2007) Journal of the American Chemical Society, 129 (31), 9566-9567 (link to paper)
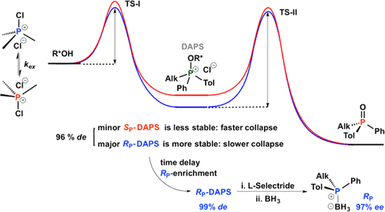
Hammond Postulate Mirroring Enables Enantiomeric Enrichment of Phosphorus Compounds via Two Thermodynamically Interconnected Sequential Stereoselective Processes
The dynamic resolution of tertiary phosphines and phosphine oxides was monitored by NMR spectroscopy. It was found that the stereoselectivity is set during the formation of the diastereomeric alkoxyphosphonium salts (DAPS), such that their initial diastereomeric excess (de) limits the final enantiomeric excess (ee) of any phosphorus products derived from them. However, 31P NMR monitoring of the spontaneous thermal decomposition of the DAPS shows consistent diastereomeric self-enrichment, indicating a higher rate constant for decomposition of the minor diastereomer. This crucial observation was confirmed by reductive trapping of the unreacted enriched DAPS with lithium tri-sec-butylborohydride (commercially distributed as L-Selectride reagent) at different time intervals after the start of reaction, which gives progressively higher ee of the phosphine product with time.
Kamalraj V. Rajendran, Kirill V. Nikitin, and Declan G. Gilheany (2015) Journal of the American Chemical Society, 137 (29), 9375–9381 (link to paper)
The dynamic resolution of tertiary phosphines and phosphine oxides was monitored by NMR spectroscopy. It was found that the stereoselectivity is set during the formation of the diastereomeric alkoxyphosphonium salts (DAPS), such that their initial diastereomeric excess (de) limits the final enantiomeric excess (ee) of any phosphorus products derived from them. However, 31P NMR monitoring of the spontaneous thermal decomposition of the DAPS shows consistent diastereomeric self-enrichment, indicating a higher rate constant for decomposition of the minor diastereomer. This crucial observation was confirmed by reductive trapping of the unreacted enriched DAPS with lithium tri-sec-butylborohydride (commercially distributed as L-Selectride reagent) at different time intervals after the start of reaction, which gives progressively higher ee of the phosphine product with time.
Kamalraj V. Rajendran, Kirill V. Nikitin, and Declan G. Gilheany (2015) Journal of the American Chemical Society, 137 (29), 9375–9381 (link to paper)
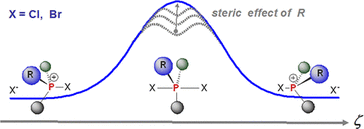
Degenerate Nucleophilic Substitution in Phosphonium Salts
Rates and energy barriers of degenerate halide substitution on tetracoordinate halophosphonium cations have been measured by NMR techniques (VT and EXSY) using a novel experimental design whereby a chiral substituent (sBu) lifts the degeneracy of the resultant salts. Concomitantly, a viable computational approach to the system was developed to gain mechanistic insights into the structure and relative stabilities of the species involved. Both approaches strongly suggest a two-step mechanism of formation of a pentacoordinate dihalophosphorane via backside attack followed by dissociation, resulting in inversion of configuration at phosphorus. The experimentally determined barriers range from <9 kcal mol–1to nearly 20 kcal mol–1, ruling out a mechansm via Berry pseudorotation involving equatorial halides. In all cases studied, epimerization of chlorophosphonium chlorides has a lower energy barrier (by 2 kcal mol–1) than the analogous bromo salts.
Elizabeth V. Jennings, Kirill Nikitin, Yannick Ortin, Declan G. Gilheany (2014) Journal of the American Chemical Society, 136 (46), 16217–16226 (link to paper)
Rates and energy barriers of degenerate halide substitution on tetracoordinate halophosphonium cations have been measured by NMR techniques (VT and EXSY) using a novel experimental design whereby a chiral substituent (sBu) lifts the degeneracy of the resultant salts. Concomitantly, a viable computational approach to the system was developed to gain mechanistic insights into the structure and relative stabilities of the species involved. Both approaches strongly suggest a two-step mechanism of formation of a pentacoordinate dihalophosphorane via backside attack followed by dissociation, resulting in inversion of configuration at phosphorus. The experimentally determined barriers range from <9 kcal mol–1to nearly 20 kcal mol–1, ruling out a mechansm via Berry pseudorotation involving equatorial halides. In all cases studied, epimerization of chlorophosphonium chlorides has a lower energy barrier (by 2 kcal mol–1) than the analogous bromo salts.
Elizabeth V. Jennings, Kirill Nikitin, Yannick Ortin, Declan G. Gilheany (2014) Journal of the American Chemical Society, 136 (46), 16217–16226 (link to paper)
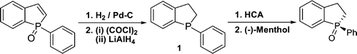
Synthesis of 2,3-Dihydro-1-phenylbenzo[b]phosphole (1-Phenylphosphindane) and Its Use as a Mechanistic Test in the Asymmetric Appel Reaction: Decisive Evidence against Involvement of Pseudorotation in the Stereoselecting Step
Racemic 2,3-dihydro-1-phenylbenzo[b]phosphole was obtained by reduction of 1-phenylbenzo[b]phosphole-1-oxide, itself derived by ring-closing metathesis of phenylstyrylvinylphosphine oxide. The title compound was then reoxidized under asymmetric Appel conditions. Comparison of the sense and degree of the stereoselectivity to those obtained with an open-chain analogue indicated that the ring system does not affect the selectivity of the process. This in turn strongly suggests that the stereoselection is not related to pseudorotamer preferences in putative phosphorane intermediates.
Damien J. Carr, Jaya Satyanarayana Kudavalli, Katherine S. Dunne, Helge Müller-Bunz, Declan G. Gilheany (2013) Journal of Organic Chemistry, 78 (20), 10500-10505 (link to paper)
Racemic 2,3-dihydro-1-phenylbenzo[b]phosphole was obtained by reduction of 1-phenylbenzo[b]phosphole-1-oxide, itself derived by ring-closing metathesis of phenylstyrylvinylphosphine oxide. The title compound was then reoxidized under asymmetric Appel conditions. Comparison of the sense and degree of the stereoselectivity to those obtained with an open-chain analogue indicated that the ring system does not affect the selectivity of the process. This in turn strongly suggests that the stereoselection is not related to pseudorotamer preferences in putative phosphorane intermediates.
Damien J. Carr, Jaya Satyanarayana Kudavalli, Katherine S. Dunne, Helge Müller-Bunz, Declan G. Gilheany (2013) Journal of Organic Chemistry, 78 (20), 10500-10505 (link to paper)
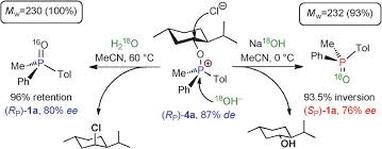
Turning Regioselectivity into Stereoselectivity: Efficient Dual Resolution of P-Stereogenic Phosphine Oxides through Bifurcation of the Reaction Pathway of a Common Intermediate
Synthetic routes that provide facile access to either enantiomeric form of a target compound are particularly valuable. The crystallization-free dual resolution of phosphine oxides that gives highly enantioenriched materials (up to 94% ee) in excellent yields is reported. Both enantiomeric oxides have been prepared from a single intermediate, (RP)-alkoxyphosphonium chloride, which is formed in the course of a selective dynamic kinetic resolution using a single enantiomer of menthol as the chiral auxiliary. The origin of the dual stereoselectivity lies in bifurcation of the reaction pathway of this intermediate, which works as a stereochemical railroad switch. Under controlled conditions, Arbuzov-type collapse of this intermediate proceeds through CO bond fission with retention of the configuration at the phosphorus center. Conversely, alkaline hydrolysis of the PO bond leads to the opposite SP enantiomer.
Kirill Nikitin, Kamalraj V. Rajendran, Helge Mller-Bunz, Declan G. Gilheany (2014) Angewandte Chemie International Edition, 53, 1906-1909 (link to paper)
Synthetic routes that provide facile access to either enantiomeric form of a target compound are particularly valuable. The crystallization-free dual resolution of phosphine oxides that gives highly enantioenriched materials (up to 94% ee) in excellent yields is reported. Both enantiomeric oxides have been prepared from a single intermediate, (RP)-alkoxyphosphonium chloride, which is formed in the course of a selective dynamic kinetic resolution using a single enantiomer of menthol as the chiral auxiliary. The origin of the dual stereoselectivity lies in bifurcation of the reaction pathway of this intermediate, which works as a stereochemical railroad switch. Under controlled conditions, Arbuzov-type collapse of this intermediate proceeds through CO bond fission with retention of the configuration at the phosphorus center. Conversely, alkaline hydrolysis of the PO bond leads to the opposite SP enantiomer.
Kirill Nikitin, Kamalraj V. Rajendran, Helge Mller-Bunz, Declan G. Gilheany (2014) Angewandte Chemie International Edition, 53, 1906-1909 (link to paper)
Air-stable primary phosphines: their synthesis and insight into their stability: By pure serendipity, we discovered that primary phosphines are not at all as air-sensitive as commonly thought. They merely have to have a HOMO higher in energy than the P lone pair. For example b-naphthyl phosphine is no more air sensitive than a moderately oxidisable aldehyde (Publication P9). This work also was performed with a talented co-worker, Dr. Lee Higham, who has now taken a position in Newcastle University and I am delighted that he was able to take over and pursue this very interesting and potentially valuable chemistry.
Publication:
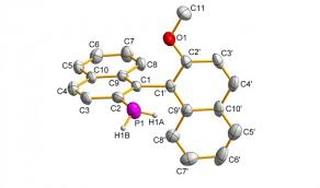
Taming a Functional Group: Creating Air-Stable, Chiral Primary Phosphanes
Enantiopure primary phosphanes have been developed which are remarkably stable to air oxidation in both solid and solution states (see example). This new class of stable ligand synthon appeas to owe its stability to conjugation in their aryl backbones with the extended π ring system. Even one extra ring is good enough.
Rachel M. Hiney, Lee J. Higham, Helge Muller-Bunz, Declan G. Gilheany (2006) Angewandte Chemie International Edition, 45 (43), 7248-7251 (link to paper)
Taming a Functional Group: Creating Air-Stable, Chiral Primary Phosphanes
Enantiopure primary phosphanes have been developed which are remarkably stable to air oxidation in both solid and solution states (see example). This new class of stable ligand synthon appeas to owe its stability to conjugation in their aryl backbones with the extended π ring system. Even one extra ring is good enough.
Rachel M. Hiney, Lee J. Higham, Helge Muller-Bunz, Declan G. Gilheany (2006) Angewandte Chemie International Edition, 45 (43), 7248-7251 (link to paper)