Deoxygenation of Phosphoryl Compounds
In the dynamic resolution work, we had discovered the new go-to reaction for this previously troublesome transformation of 3˚/2˚ phosphine oxides (Chem. Commun. 2012, 48, 817). Then, because it relied on a chloro leaving group, I hypothesised that it could be extendable to aminophosphine oxides where the N would be less likely to leave.
Selected publications:
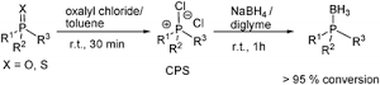
Simple unprecedented conversion of phosphine oxides and sulfides to phosphine boranes using sodium borohydride
A variety of phosphine oxides and sulfides can be efficiently converted directly to the corresponding phosphine boranes using oxalyl chloride followed by sodium borohydride. Optically active P-stereogenic phosphine oxides can be converted stereospecifically to phosphine boranes with inversion of configuration by treatment with Meerwein's salt followed by sodium borohydride.
Kamalraj V. Rajendran, Declan G. Gilheany (2012) Chemical Communications, 48, 817-819 (link to paper)
A variety of phosphine oxides and sulfides can be efficiently converted directly to the corresponding phosphine boranes using oxalyl chloride followed by sodium borohydride. Optically active P-stereogenic phosphine oxides can be converted stereospecifically to phosphine boranes with inversion of configuration by treatment with Meerwein's salt followed by sodium borohydride.
Kamalraj V. Rajendran, Declan G. Gilheany (2012) Chemical Communications, 48, 817-819 (link to paper)
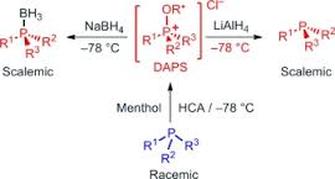
A U-Turn in the Asymmetric Appel Reaction: Stereospecific Reduction of Diastereomerically Enriched Alkoxyphosphonium Salts Allows the Asymmetric Synthesis of P-Stereogenic Phosphanes and Phosphane Boranes
An efficient one-pot synthesis has been developed of enantioenriched P-stereogenic phosphanes and phosphane boranes from the corresponding racemic phosphanes in excellent yield under asymmetric Appel conditions. The chiral auxiliary (menthol) can also be recovered unchanged. The simple and efficient protocol significantly expands the scope of our asymmetric Appel process. The crucial step in the preparation involves stereospecific reduction of intermediate diastereomeric alkoxyphosphonium salts, which are obtained in the reaction of phosphane, hexachloroacetone, and menthol. Thereby, reaction with LiAlH4 or NaBH4 gives the corresponding phosphanes or phosphane boranes, respectively.
Kamalraj V. Rajendran, Jaya S. Kudavalli, Katherine S. Dunne, Declan G. Gilheany (2012) European Journal of Organic Chemistry, 2012 (14),2720-2723 (link to paper)
An efficient one-pot synthesis has been developed of enantioenriched P-stereogenic phosphanes and phosphane boranes from the corresponding racemic phosphanes in excellent yield under asymmetric Appel conditions. The chiral auxiliary (menthol) can also be recovered unchanged. The simple and efficient protocol significantly expands the scope of our asymmetric Appel process. The crucial step in the preparation involves stereospecific reduction of intermediate diastereomeric alkoxyphosphonium salts, which are obtained in the reaction of phosphane, hexachloroacetone, and menthol. Thereby, reaction with LiAlH4 or NaBH4 gives the corresponding phosphanes or phosphane boranes, respectively.
Kamalraj V. Rajendran, Jaya S. Kudavalli, Katherine S. Dunne, Declan G. Gilheany (2012) European Journal of Organic Chemistry, 2012 (14),2720-2723 (link to paper)
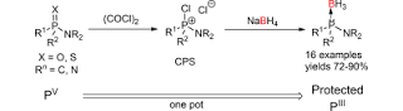
Cleavage of P=O in the Presence of P–N: Aminophosphine Oxide Reduction with In Situ Boronation of the PIII Product
In contrast to tertiary phosphine oxides, the deoxygenation of aminophosphine oxides is effectively impossible due to the need to break the immensely strong and inert P=O bond in the presence of a relatively weak and more reactive P-N bond. This long-standing problem in organophosphorus synthesis is solved by use of oxalyl chloride, which chemoselectively cleaves the P=O bond forming a chlorophosphonium salt, leaving the P-N bond(s) intact. Subsequent reduction of the chlorophosphonium salt with sodium borohydride forms the PIII aminophosphine borane adduct. This simple one-pot procedure was applied with good yields for a wide range of P-N containing phosphoryl compounds. The borane product can be easily deprotected to produce the free PIII aminophosphine. Along with no observed P-N bond cleavage, the use of sodium borohydride also permits the presence of ester functional groups in the substrate. The availability of this methodology opens up previously unavailable synthetic options in organophosphorus chemistry, two of which are exemplified.
Niall P. Kenny, Kamalraj V. Rajendran, Elizabeth V. Jennings, Declan G. Gilheany (2013) Chemistry: A European Journal, 19(42),14210-14214 (link to paper)
In contrast to tertiary phosphine oxides, the deoxygenation of aminophosphine oxides is effectively impossible due to the need to break the immensely strong and inert P=O bond in the presence of a relatively weak and more reactive P-N bond. This long-standing problem in organophosphorus synthesis is solved by use of oxalyl chloride, which chemoselectively cleaves the P=O bond forming a chlorophosphonium salt, leaving the P-N bond(s) intact. Subsequent reduction of the chlorophosphonium salt with sodium borohydride forms the PIII aminophosphine borane adduct. This simple one-pot procedure was applied with good yields for a wide range of P-N containing phosphoryl compounds. The borane product can be easily deprotected to produce the free PIII aminophosphine. Along with no observed P-N bond cleavage, the use of sodium borohydride also permits the presence of ester functional groups in the substrate. The availability of this methodology opens up previously unavailable synthetic options in organophosphorus chemistry, two of which are exemplified.
Niall P. Kenny, Kamalraj V. Rajendran, Elizabeth V. Jennings, Declan G. Gilheany (2013) Chemistry: A European Journal, 19(42),14210-14214 (link to paper)